O que é atrofia muscular espinhal (AME)

A atrofia muscular espinhal (AME) é uma grave doença degenerativa geneticamente determinada, que se caracteriza pela perda progressiva dos motoneurônios alfa localizados no corno anterior da medula espinhal e na coluna eferente somática de núcleos de nervos cranianos situados no tronco encefálico. Essa degeneração dos motoneurônios leva a fraqueza muscular proximal progressiva, atrofia dos músculos esqueléticos e graus variáveis de insuficiência ventilatória. A forma mais comum de AME é causada por mutações no gene de sobrevida do neurônio motor 1 (survival motor neuron 1-SMN1), e é herdada em um padrão autossômico recessivo.
Aproximadamente 95% a 98% dos indivíduos com diagnóstico clínico de AME apresentam deleção do éxon 7 em ambas as cópias do gene SMN1, enquanto cerca de 2% a 5% dos pacientes apresentam deleção do éxon 7 em um dos alelos do gene e uma mutação intragênica no outro alelo. (Boletim ABNEWS 2019)Dr. Edmar Zanoteli, professor do Departamento de Neurologia da Faculdade de Medicina da Universidade de São Paulo e coordenador do DC de Moléstias Neuromusculares da ABN(Academia Brasileira de Neurologia)

Johann Hoffmann (1857 – 1919) foi um neurologista alemão.
A descoberta da AME
A AME foi descrita pela primeira vez por um cientista alemão chamado Johann Hoffman e um cientista austríaco chamado Guido Werdnig. Os dois homens notaram vários casos de bebês que desenvolveram fraqueza muscular nos primeiros meses de vida. Eles também notaram que essa condição parecia ser familiar.
Enquanto investigavam essa condição misteriosa, eles viram que as células do neurônio motor nesses bebês pareciam degenerar, especificamente em uma seção da medula espinhal conhecida como corno anterior. Esta seção está localizada na parte frontal da medula espinhal e está associada aos músculos esqueléticos – ao contrário de outras seções da medula espinhal, que estão associadas ao tato e outros sentidos.
Suas observações levaram à identificação de atrofia muscular espinhal. Até hoje, a AME tipo 1 costuma ser chamada de doença de Werdnig-Hoffman, em reconhecimento a esses dois cientistas.
Algumas décadas depois, dois outros cientistas, Erik Kugelberg e Lisa Welander, foram capazes de diferenciar as formas de início tardio de AME de outras doenças semelhantes, como distrofia muscular. É por isso que a AME tipo 3 também é chamada de doença de Kugelberg-Welander.
Johann Hoffmann (1857 – 1919) foi um neurologista alemão.

Guido Werdnig (1844 – 1919) foi um neurologista austríaco.
Da descoberta ao tratamento
Drs. Werdnig e Hoffman fizeram sua descoberta pela primeira vez na década de 1890, mas seria mais um século antes que a próxima grande descoberta na AME fosse feita. Durante os anos 1900, os cientistas fizeram grandes avanços em nossa compreensão da genética humana, incluindo a descoberta do DNA. Essas descobertas ajudaram a aumentar nosso conhecimento sobre muitas doenças, incluindo AME.
Em 1995, a Dra. Judith Melki e sua equipe identificaram e determinaram a composição do DNA do gene 1 do neurônio motor de sobrevivência (SMN1) . Eles demonstraram que, em indivíduos com AME, esse gene foi deletado ou sofreu mutação. O Dr. Melki também identificou o gene SMN2, o “gene reserva” da AME. Quatro anos depois, em 1999, o Dr. Umrao Monani no laboratório de Arthur Burghes e o Dr. Chris Lorson no laboratório de Elliot Androphy descreveram o erro de emenda no SMN2. Este trabalho foi financiado pela Cure SMA.
A descoberta do SMN1 abriu as portas para um teste genético para confirmar o diagnóstico de AME. Antes disso, o diagnóstico era feito com base nos sinais e sintomas visíveis da AME.
As descobertas do SMN1 e SMN2 também abriram as portas para a descoberta de novos tratamentos. Em 2003, apenas alguns anos após os drs. Monani e Lorson identificaram o erro de splicing no SMN2, o Cure SMA concedeu a primeira bolsa de pesquisa ao Dr. Ravindra Singh no laboratório de Elliot Androphy para estudar uma abordagem que consertaria o splicing do SMN2.
Essa abordagem acabou levando a descoberta e o desenvolvimento do Spinraza por Frank Bennet na Ionis e Adrian Krainer no Cold Spring Harbor Laboratory, que foi aprovado pelo FDA em dezembro de 2016 e comunidade da atrofia muscular espinhal comemorou a aprovação do primeiro tratamento que visa a causa genética subjacente da AME. Este foi um marco histórico de mais de 100 anos, possibilitado por pesquisadores dedicados e pela comunidade que apoiou seu trabalho.
Também em 2016, Cure SMA homenageou Judith Melki na Conferência Anual da AME, por suas contribuições importantes para a nossa compreensão da AME. A cada ano, a Conferência Anual da AME reúne centenas de cientistas que estão investigando novas questões sobre a AME. Eles estão construindo sobre as descobertas dos drs. Werdnig, Hoffman e Melki, na esperança de desenvolver mais tratamentos para todos os tipos, idades e estágios de AME.
Nossos agradecimentos aos pesquisadores inteligentes e determinados que trabalham todos os dias em nome de nossa comunidade.
SMA = AME em inglês.
CURE SMA = Associação dos Estados Unidos pioneira na luta contra a AME.
Fonte: www.curesma.org

Dra. Judith Melki

Dr. Ravindra Singh

Dr. Frank Bennet
Hereditariedade
A HEREDITARIEDADE DA ATROFIA MUSCULAR ESPINHAL (AME)
A Atrofia Muscular Espinhal (AME) 5q é uma doença genética de herança autossômica recessiva, o que significa que, para apresentar os sintomas da doença, os indivíduos devem possuir dois alelos SMN1 com alteração, um proveniente do pai e outro da mãe, na maioria dos casos.
O pai e a mãe, que possuem uma cópia do alelo alterado, são denominados portadores e não apresentam a doença. A chance que estes pais tenham um filho ou filha afetado é de 25% em cada gravidez. Pessoas com casos de Atrofia Muscular Espinhal (AME) ou outras doenças neuromusculares na família devem ser encaminhadas ao aconselhamento genético. Além disso, é essencial encaminhar a família para o aconselhamento genético. O aconselhamento é um processo de fornecer às pessoas com Atrofia Muscular Espinhal (AME) e seus familiares não somente informações sobre a doença, mas também orientações que englobem o planejamento familiar. Dentro desse planejamento, informações sobre o risco de ocorrência ou recorrência da AME são fornecidas, isto é, quais as chances de uma pessoa com Atrofia Muscular Espinhal (AME)ter um filho(a) afetado(a) pela doença ou de um casal ter um(a) filho(a) com a Atrofia Muscular Espinhal (AME); o mesmo vale para familiares com histórico da doença na família.
Devem ser discutidas todas as opções, incluindo triagem de portadores no casal, diagnóstico pré-natal do feto, doação de esperma ou óvulo e fertilização in vitro com diagnóstico pré-implantacional, a fim de ajudar os indivíduos a tomarem decisões médicas e pessoais informadas.


Tipos de AME
Não há duas pessoas com AME que tenham experiências idênticas.
Mesmo entre aqueles com o mesmo tipo, a vivência da doença pode ser diferente. As decisões sobre cuidados e tratamento devem ser tomadas de acordo com as necessidades de cada indivíduo.
Existem cinco tipos de atrofia muscular espinhal (AME): Tipos 0, 1, 2, 3 e 4. O tipo de AME é baseado na idade em que os sintomas começam e no marco físico mais alto alcançado. Mesmo dentro de cada tipo, as habilidades podem variar de pessoa para pessoa. Além disso, os indivíduos com AME podem perder a função com o tempo se os músculos continuarem a enfraquecer.
Com o tratamento, os indivíduos podem obter mais marcos físicos do que teriam de outra forma. E, à medida que a triagem neonatal para AME se torna mais comum, os bebês podem receber tratamento
antes mesmo do início dos sintomas. Por causa desses fatores, os médicos acreditam que logo poderemos parar de descrever a AME como “tipos” específicos e, em vez disso, nos concentrarmos no marco motor mais alto alcançado: não-babá, babá e andador. Mas, mesmo assim, ainda haverá uma ampla gama de gravidade associada à AME.
AME tipo 0 é muito raro e muito grave. Os sintomas começam antes do nascimento e são vistos como diminuição do movimento fetal nas semanas anteriores ao parto. Ao nascer, o bebê apresenta fraqueza severa e frequentemente dificuldade para respirar, alimentar-se e pode apresentar contraturas nas articulações e defeitos cardíacos. Esses bebês geralmente requerem suporte respiratório e alimentar antes de confirmar o diagnóstico. Esses bebês podem sobreviver alguns meses.
Dificuldades respiratórias: Frequentemente necessitam de suporte ventilatório nos primeiros minutos ou horas após o nascimento.Dificuldades motoras: hipotonia profunda, fraqueza grave e contraturas articulares. Dificuldades de alimentação: apresentam grave disfagia e incapacidade de sucção para mamar.
Expectativa de vida: a grande maioria dos pacientes acaba falecendo nos primeiros dias ou semanas de vida, em geral não ultrapassando seis meses de idade.
Também conhecida como doença de Werdnig-Hoffmann, a AME tipo 1 é a forma mais comum (60%) e grave, geralmente diagnosticada durante os primeiros 6 meses de uma criança. Os bebês com AME tipo 1 enfrentam muitos desafios físicos, incluindo fraqueza muscular e dificuldade para respirar, tossir e engolir. Eles podem precisar de assistência respiratória ou de um tubo de alimentação. Se não for tratado, o tipo 1 pode ser fatal no início da vida.
O tipo 2 geralmente é diagnosticado após os 6 meses de idade, mas antes dos 2 anos de idade. O primeiro sinal geralmente é um atraso no cumprimento dos marcos do motor ou falha em cumprir os marcos totalmente. Os indivíduos com AME tipo 2 geralmente podem sentar-se sem ajuda, embora possam precisar de ajuda para se sentarem, mas são incapazes de andar e precisam de uma cadeira de rodas.
Também chamada de doença de Kugelberg-Welander ou AME juvenil, o tipo 3 geralmente é diagnosticado após os 18 meses de idade, mas antes dos 3 anos de idade. No entanto, o AME tipo 3 pode ser diagnosticado na adolescência. Indivíduos com AME tipo 3 são inicialmente capazes de andar, mas têm mobilidade cada vez mais limitada à medida que crescem e, eventualmente, muitos precisam usar uma cadeira de rodas.
AME tipo 4 é muito raro, menos de 1% de todos os casos diagnosticados. Geralmente surge na idade adulta e leva a um leve comprometimento motor. Embora os sintomas possam começar aos 18 anos, geralmente começam após os 35 anos.
Outras Formas de AME
Ao contrário dos tipos primários de AME descritos acima, essas outras formas de AME são causadas por mutações em genes diferentes do gene SMN1.
AME dificuldade respiratória (SMARD)
SMARD é uma forma muito rara de AME tipo 1 que afeta a medula espinhal superior mais do que a medula espinhal inferior. Bebês com SMARD apresentam dificuldade respiratória severa e fraqueza nos braços e músculos próximos. SMARD é causado por uma mutação específica e pode ser diagnosticado por meio de testes genéticos.
Distal AME
AME distal é outra forma rara de AME. Ao contrário de outras formas de AME, a AME distal pode ser herdada de apenas um dos pais. A fraqueza da AME distal afeta as mãos e os pés.
Doença de Kennedy
Outra forma rara de AME, a doença de Kennedy é uma doença genética ligada ao X, o que significa que afeta apenas homens. Geralmente aparece entre as idades de 30 e 50 anos. Causa fraqueza e atrofia muscular em todo o corpo, que é mais perceptível nas pernas e nos braços. Também é especialmente perceptível no rosto e na garganta, e causa dificuldades na fala e na deglutição e grandes cãibras musculares.
Fonte: www.curesma.org
Diagnóstico
Após a suspeita clínica de Atrofia Muscular Espinhal (AME), o paciente deve ser encaminhado à realização do teste genético para diagnóstico, tendo em vista que a doença é genética. O exame é feito por meio de coleta de sangue, de saliva ou raspado de bochecha para a extração de DNA e análise quantitativa do gene SMN1.
Atualmente, essas análises são feitas por meio dos métodos MLPA (do inglês Multiplex Ligation-Dependent Probe Amplification), PCR quantitativa (do inglês Polymerase Chain Reaction) quantitativa ou NGS (do inglês Next Generation Sequencing).
O algoritmo diagnóstico é mostrado na figura abaixo. Apesar de alguns testes também propiciarem a quantificação do número de cópias do SMN2, esse dado não é necessário para diagnóstico, mas é importante como complemento para auxiliar na predição da gravidade da doença.
Atualmente é possível realizar o exame de DNA para confirmação de diagnóstico através do programa AME+(mais), para maiores informações você pode entrar em contato pelo telefone 0800 200 0550 ou pelo email: amemais@biogen.com
Fonte:https://www.juntospelaame.com.br


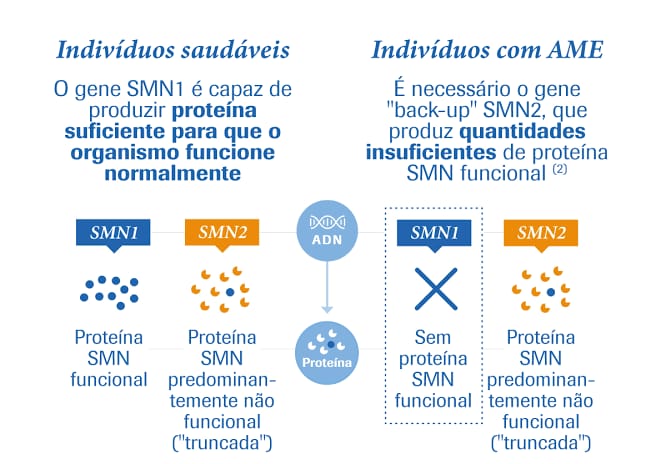
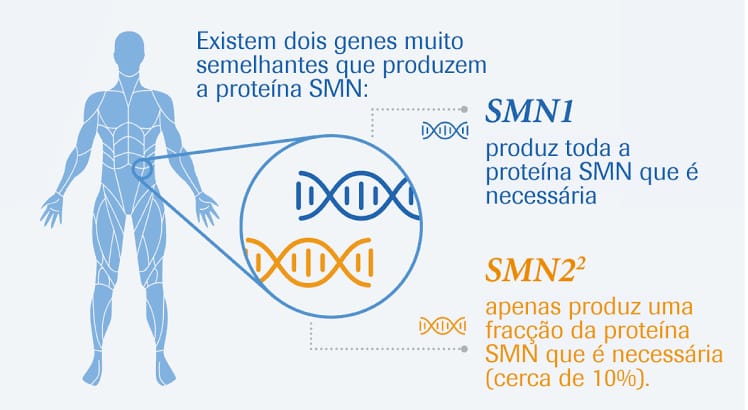
O que é o gene SMN2 e qual a diferença quando comparado ao gene SMN1?
O que é o gene SMN2 e qual a diferença quando comparado ao gene SMN1? O gene SMN2 é muito parecido com o gene SMN1, mas não é capaz de produzir a mesma quantidade da proteína SMN. Os pacientes com AME apresentam mutação no gene SMN1, mas retêm o gene SMN2. No entanto, a presença do gene SMN2 não é suficiente para evitar a doença, pois a quantidade de proteína produzida por esse gene é pequena. Alguns pacientes têm mais cópias do gene SMN2, então produzem uma quantidade maior da proteína SMN e, portanto, apresentam um quadro clínico mais leve. Em geral, os pacientes com AME tipo 1 apresentam duas cópias do gene SMN2; aqueles com AME tipo 2 apresentam, usualmente, três cópias do gene SMN2; e os pacientes com AME tipo 3 possuem de três a
quatro cópias do gene SMN2. Sabendo disso, os pesquisadores há alguns anos têm tentado “ativar” o gene SMN2 a produzir mais proteína e melhorar o quadro clínico dos pacientes. Vários medicamentos já foram testados, embora muitos deles não tenham funcionado
adequadamente. Por outro lado, medicamentos como a nusinersena (Spinraza® – Biogen Brasil Produtos Farmacêuticos Ltda.) e as moléculas pequenas têm demonstrado resultados promissores.
Qual a importância do diagnóstico genético precoce?
 Quanto mais precoce o início dos tratamentos melhor são os efeitos dos medicamentos. Assim, o diagnóstico genético deve ser feito o mais precocemente possível. Em alguns países já tem sido realizada, inclusive, triagem neonatal na tentativa de identificar indivíduos em fase pré-sintomática. O teste genético, além de determinar se o paciente tem a mutação no gene SMN1, também vai indicar o número de cópias do gene SMN2, e essa informação é importante para dar alguma sugestão do prognóstico.
Quanto mais precoce o início dos tratamentos melhor são os efeitos dos medicamentos. Assim, o diagnóstico genético deve ser feito o mais precocemente possível. Em alguns países já tem sido realizada, inclusive, triagem neonatal na tentativa de identificar indivíduos em fase pré-sintomática. O teste genético, além de determinar se o paciente tem a mutação no gene SMN1, também vai indicar o número de cópias do gene SMN2, e essa informação é importante para dar alguma sugestão do prognóstico.


Teste do Pezinho será ampliado e detectará até 50 novas doenças.
Foi sancionada a Lei nº 14.154, que amplia para 50 o número de doenças rastreadas pelo Teste do Pezinho oferecido pelo Sistema Único de Saúde (SUS). O exame, feito por meio da coleta de gotas de sangue dos pés de recém-nascidos, atualmente engloba apenas seis doenças.
As seis doenças abrangidas atualmente são: fenilcetonúria, hipotireoidismo congênito, síndromes falciformes, fibrose cística, hiperplasia adrenal congênita e deficiência de biotinidase. Com a nova lei, o exame passará a abranger 14 grupos de doenças. Essa ampliação ocorrerá de forma escalonada e caberá ao Ministério da Saúde estabelecer os prazos para implementação de cada etapa do processo.
Na primeira etapa da ampliação do teste está prevista a inclusão de doenças relacionadas ao excesso de fenilalanina; patologias relacionadas à hemoglobina; e toxoplasmose congênita. Na segunda etapa, serão detectados: nível elevado de galactose no sangue; aminoacidopatias; distúrbio do ciclo de ureia; e distúrbios de betaoxidação de ácidos graxos.
Na terceira etapa, serão incluídas no Teste do Pezinho oferecido pelo SUS doenças que afetam o funcionamento celular, e, na quarta etapa, problemas genéticos no sistema imunológico. A partir da quinta etapa será testada também a atrofia muscular espinhal. As mudanças entram em vigor 365 dias após a publicação da lei.
Foi sancionada a Lei nº 14.154, que amplia para 50 o número de doenças rastreadas pelo Teste do Pezinho oferecido pelo Sistema Único de Saúde (SUS). O exame, feito por meio da coleta de gotas de sangue dos pés de recém-nascidos, atualmente engloba apenas seis doenças.
As seis doenças abrangidas atualmente são: fenilcetonúria, hipotireoidismo congênito, síndromes falciformes, fibrose cística, hiperplasia adrenal congênita e deficiência de biotinidase. Com a nova lei, o exame passará a abranger 14 grupos de doenças. Essa ampliação ocorrerá de forma escalonada e caberá ao Ministério da Saúde estabelecer os prazos para implementação de cada etapa do processo.
Na primeira etapa da ampliação do teste está prevista a inclusão de doenças relacionadas ao excesso de fenilalanina; patologias relacionadas à hemoglobina; e toxoplasmose congênita. Na segunda etapa, serão detectados: nível elevado de galactose no sangue; aminoacidopatias; distúrbio do ciclo de ureia; e distúrbios de betaoxidação de ácidos graxos.
Na terceira etapa, serão incluídas no Teste do Pezinho oferecido pelo SUS doenças que afetam o funcionamento celular, e, na quarta etapa, problemas genéticos no sistema imunológico. A partir da quinta etapa será testada também a atrofia muscular espinhal. As mudanças entram em vigor 365 dias após a publicação da lei.
A Primeira-dama da República, Michelle Bolsonaro, afirmou que pessoas que possuem doenças raras (algumas delas incluídas no Teste do Pezinho) ficaram “invisíveis” durante muitos anos, mas que o Governo Federal trabalha para mudar a realidade desses pacientes. “As doenças raras atingem de 6% a 8% da população mundial. No Brasil, esse número significa por volta de 14 milhões de pessoas, e 75% dos casos manifestam-se na infância, ou seja, o diagnóstico é fundamental para salvar vidas.”

